A novel modelling strategy for simulating the structural dynamics of complex molecular systems in solutions is presented.
Photocatalytic processes are highly relevant in both academic research and in a range of industrial applications. In recent years, new powerful X-ray facilities have made it possible to observe these processes in real time. Such experiments do not provide direct insight, but need to be accompanied by extensive modelling and theory. Especially photocatalytic processes taking place in solution are challenging to monitor, as photocatalytic complexes tend to undergo structural modifications or change the solvent in which they are embedded. In the project, a novel modelling strategy for simulating the structural dynamics of complex molecular systems in solutions was developed.
Being able to observe the dynamics of the chemical bond in real time is one of the greatest achievements of physical chemistry. Nuclear vibrational motion unfolds on a very short time scale, the femtosecond time scale (1 fs = 10-15 s). Previously this was out of reach, but over the last three decades femtosecond X-ray scattering measurements have emerged. In the project, experiments were carried out as part of an experimental campaign at the Linac Coherent Light Source (LCLS) X-ray Free Electron Laser (XFEL) of Stanford University, USA.
Photocatalytic reactions involving transition metal complexes in solution are popular targets for timeresolved experiments. Apart from their scientific and industrial importance, these systems are attractive to study due to their stability in solution, remarkable photophysical properties, and the presence of electronrich atoms. However, in order to exploit their structure-function relationships an understanding of the mechanisms behind ultrafast light-induced reactions in complex environments is required. The experiments aimed to elucidate these mechanisms. These novel experiments cover grounds often dominated by complex interplays between vibrational relaxation, solvent effects and electronic couplings. Therefore, solid theoretical and modelling strategies are needed along with advanced computational methods.
In the project, a novel multiscale atomistic modelling strategy for simulating the structural dynamics of complex molecular systems was implemented and applied. The method is based on the direct Born-Oppenheimer Molecular Dynamics propagation of the nuclei and treats solvent effects within a quantum mechanics / molecular mechanics (QM/MM) framework.
Initially, the QM/MM scheme was augmented to include electronic excited states with arbitrary spin multiplicity using a ΔSCF approach.
Further, the photocatalytic diplatinum(II) complex [Pt2(P2O5H2)4]4-, abbreviated PtPOP, was studied. Owing to its nuclear and electronic structures, PtPOP is the prototype system of choice for photophysical studies within a family of highly photoreactive d⁸-d⁸ binuclear complexes which can catalytically abstract hydrogen and halogen atoms from different substrates. It was shown how ΔSCF for the first time provided computational evidence that the lowest-lying singlet and triplet excited states have parallel potential energy surfaces along the Pt-Pt coordinate. Also, the synergy between time-resolved experiments and simulations in unravelling the photoinduced ultrafast dynamics of the complex in water was highlighted.
Finally, a step forward was taken in understanding excited-state vibrational relaxation in solution. It was shown that after relaxation, PtPOP does not, as previously believed, retain the symmetry of the ground state; and excess Pt-Pt vibrational energy is first directed towards vibrational modes involving the ligands, while the role of the solvent is to favor intramolecular vibrational energy redistribution in the complex.
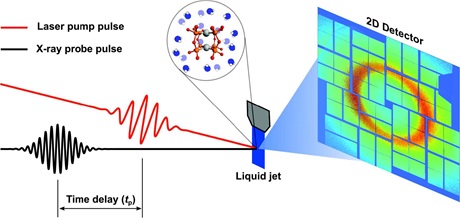
Schematical illustration of an optical pump/X-ray probe setup for time-resolved X-ray diffuse scattering (XDS) experiments.