The transformation of one set of chemical substances (reactants) to another (products) is of fundamental importance for chemistry and biology. In a crude manner, traditional means as heat and light (i.e., temperature and frequency of photons) have been employed for decades to guide the reactants into particular product channels.
The atomic-scale dynamics of molecules and chemical reactions unfolds on the femtosecond (10-15 s) timescale and is governed by the principles of quantum mechanics. These principles appear to offer the promise of a vastly improved performance over what is achievable within the classical framework of so-called incoherent processes. To that end, photochemistry with coherent laser light is a potential quantum technology that is a potentially attractive alternative to classical photochemistry.
The PhD thesis investigates the interaction between laser light and molecules in the regime of the so-called non-resonant dynamic Stark effect. This amounts to a time-dependent modification of the interaction potential determined by the pulse envelope of laser pulses and independent of laser frequency. The lack of frequency dependence is a very convenient feature for experimental implementations.
Through theory and computations, it is demonstrated how molecules can be rotationally and vibrationally excited. In a collaboration with an experimental group, the field-free rotational motion of I2 is analyzed. The experimental observations show clear signatures of so-called quadrupole coupling to the nuclear spin states and all features of this coupling are completely accounted for.
It is also shown how laser light can be used in optical purification of a racemic mixture which is a 50/50 mixture of enantiomers, i.e., molecules which are each other’s mirror images. In order to close the gap between theory and experiment, the optimized laser-light pulses are constructed in similar way as in pulse shapers that are used in experimental setups.
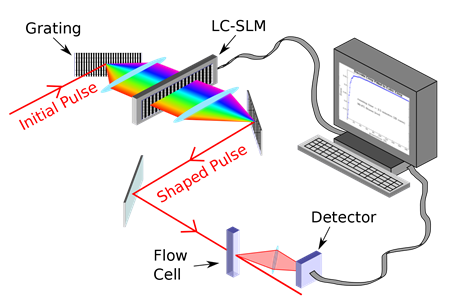
Sketch of a closed-loop control system.